
Protein language models are bad at mutational effect prediction
Biology is hard. Yes, even for AI.
Last summer, I wrote a post claiming that protein language models (pLMs) showed poor performance on viral data. At the time, this was a preliminary result based on a handful of datasets, and I said as much. I also said we were going to do more work on this problem. Well, we have done the work now, and I can confidently say that protein language models perform worse on viral proteins than on cellular proteins. However, more importantly, they perform poorly on either, when the task considered is mutational effect prediction (i.e., predicting by how much a mutation changes the activity or fitness1 of a protein). The paper is on bioRxiv.2
If you’re following the literature on pLMs, you may be confused by my statement. There are many papers that seemingly show excellent performance. In fact, whenever a new pLM is released, one of the standard benchmarking tasks is typically mutational effects prediction. And performance often appears to be excellent. Unfortunately, much of this apparent success is just people confusing themselves over what is actually happening. If you dig deeper you can find the cracks under the surface.
Before I continue, let me quickly specify what kind of modeling situation I’m referring to. I’m specifically focusing on supervised learning of mutational effects from deep mutational scanning (DMS) data. In this situation, we have experimental data for thousands of mutants of a protein, we split the data into training and test sets, train the model on the training set, and then evaluate on the test set. This is distinct from so-called zero-shot predictions, which are also popular, where we don’t have a training set and just predict mutational effects from the pre-trained model, without learning anything about the specific dataset at hand. Zero-shot predictions have their own issues. I’ll not discuss them here. Everything in this post is exclusively about supervised learning.
The biggest problem in supervised learning is data leakage, where information from the training set leaks into the test set, and this is definitely happening in the field of mutational effects prediction. The problem is that it is common to treat the thousands of mutations in a DMS dataset as independent from each other (Figure 1A, pooled split), ignoring the fact that there will often be multiple mutations at the same site and those mutations will have correlated effects. Thus, the model can learn which sites in a protein are sensitive to mutations and which are not and make predictions based on this information rather than on the specific biochemistry of individual mutations. To avoid this data leakage problem, we have to stratify by site when generating training–test splits (Figure 1A, stratified by site).
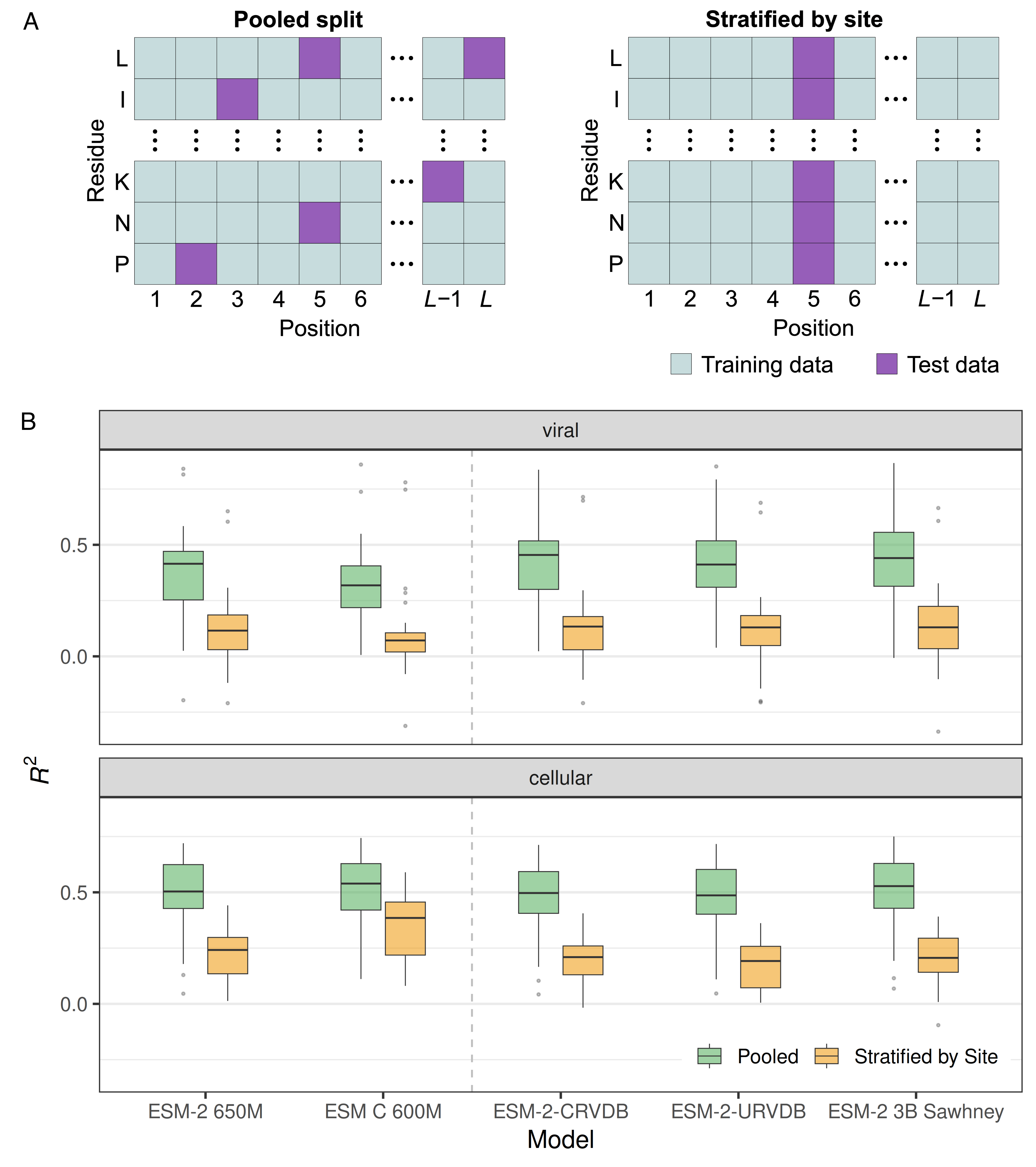
When comparing models trained and evaluated either on pooled splits or on splits stratified by site, we see a huge drop in performance in the latter (Figure 1B). And this drop exists regardless of whether we are working with viral or cellular proteins, and whether we’re using a generic model or one fine-tuned for viral data. In fact, most models show roughly the same performance. All models perform poorly on site-stratified data.3
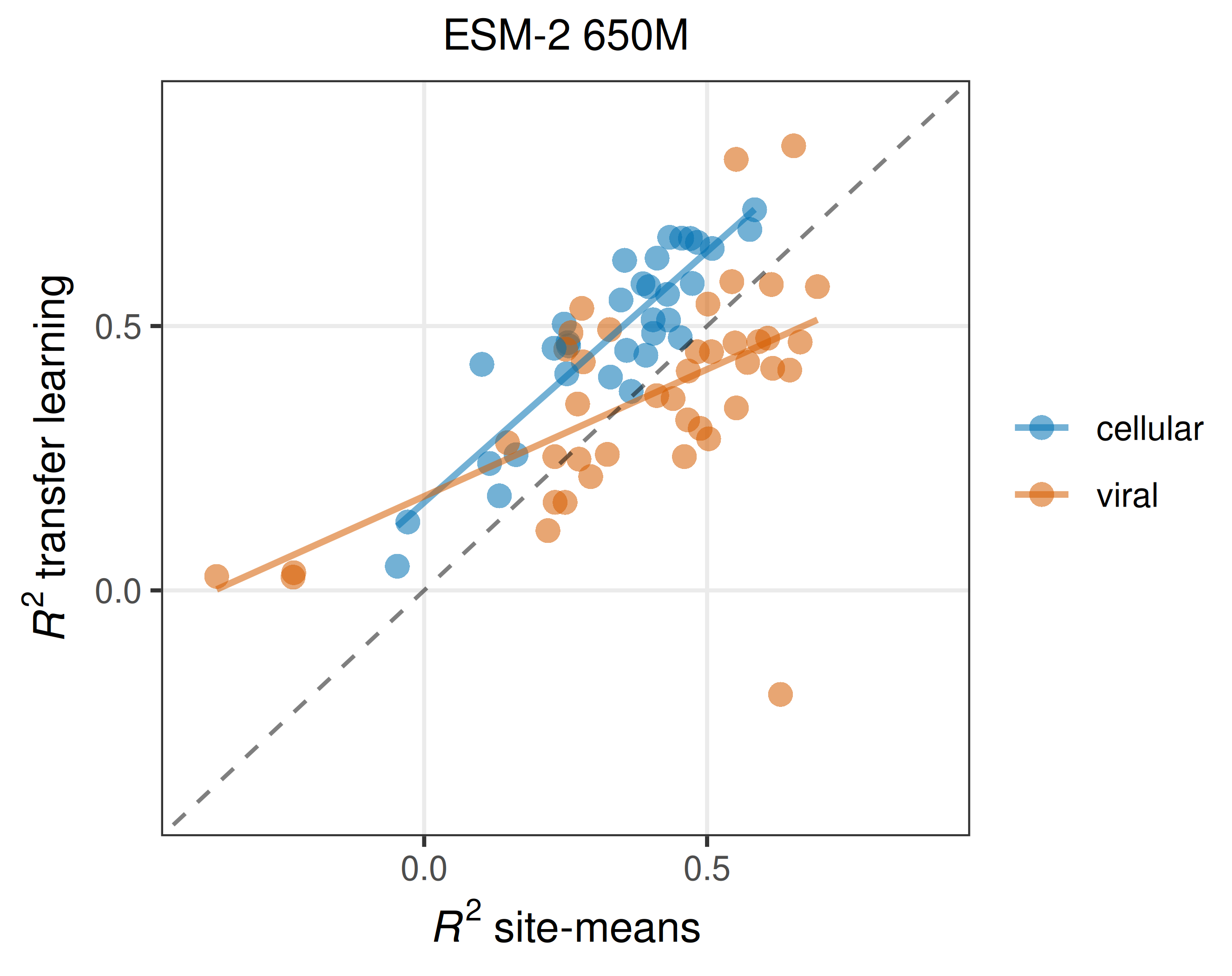
Now you may wonder how correlated different mutations at the same site really are. There’s a simple way to find out: For pooled splits, we can simply take the average fitness effect in the training data at each site and use this as our prediction for the test data. I want to emphasize how simplistic of a model this is: We are simply saying that any unseen mutation is going to have the average effect at its site. How well does such a model perform? On cellular data, almost as well as a full-scale protein language model, and on viral data, better than a full-scale protein language model! In Figure 2, dots above the dashed line imply that the pLM is better, and dots below the dashed line imply that simple site means are better. You can see how for more than half of the viral datasets, site means are better than the pLM. And for cellular datasets, even though all the blue dots are above the dashed line, they are only shifted upwards by a small amount. If the site-means model does well, the pLM also does well (and a little better than the site-means model), and if the site-means model doesn’t do well the pLM also doesn’t do well (but it still does a little better than the site-means model). I think this is a devastating result. In the vast majority of cases, pLMs with millions of parameters do barely better than a model that just memorizes mean effects at each site.
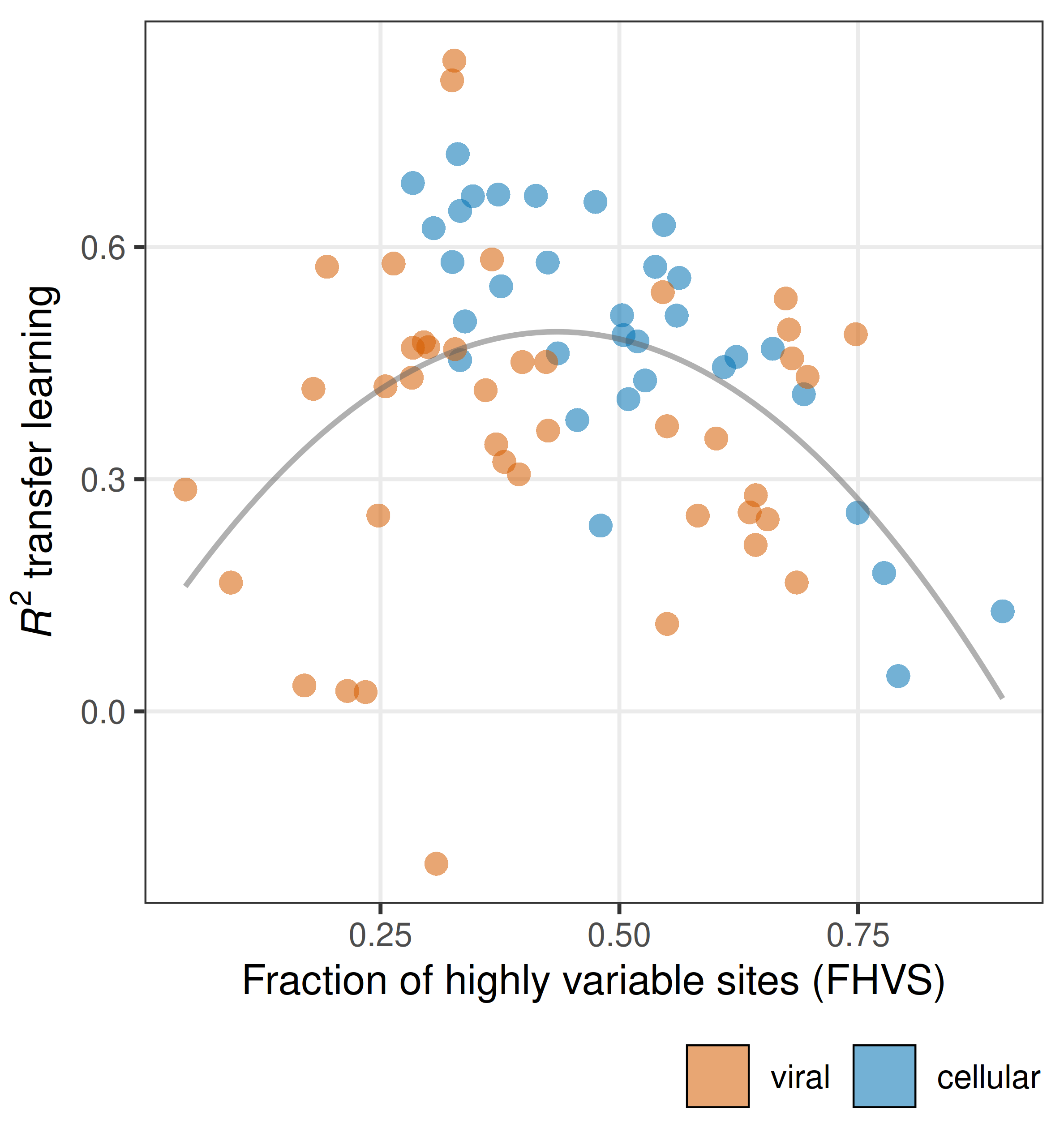
Another obvious take-away from Figure 2 is the variation in model performance across datasets is huge. For some datasets predictions are apparently very easy, and for other datasets predictions are nearly impossible. We spent a lot of effort trying to understand what makes a dataset predictable. In brief, it comes down to variation within and among sites.4 In particular, models only do well on datasets that have an intermediate fraction of highly variable sites (Figure 3). There are no datasets with either a very low or a very high fraction of variable sites for which model performance is good. Interestingly, the viral and the cellular proteins separate on this dimension. Many of the viral datasets for which prediction is difficult have a particularly low fraction of highly variable sites, and many of the cellular datasets for which prediction is difficult have a particularly high fraction of highly variable sites. This may be one of the main reasons why predictions on viral and cellular datasets differ.
So, we have learned that apparent good pLM performance on mutational effects prediction is largely driven by site effects (knowing the average fitness at a site allows you to make pretty good predictions for new mutations at that site), and these site effects can leak into the test data when using pooled splits. Moreover, there are aspects that are intrinsic to the dataset and completely independent of the model that determine how well a model will perform. These are related to the fitness variation within and among sites. Datasets with just the right fitness distribution are highly predictable (even by bad models) and datasets with the wrong fitness distribution can’t be predicted by any models. The relative difference in performance between different models is comparatively minor.
One last issue is the metric used to assess model performance. We use R2 to measure performance, when most other studies use Spearman ρ. I’m not a big fan of ρ. I think it artificially inflates perceived model performance. First, all else being equal, and even though both are always less than or equal to one, ρ will always be larger than R2. This means ρ has less discriminatory power. An excellent model and a good model may have quite similar ρ values even though their R2 values are not that similar. A ρ = 0.8 and a ρ = 0.6 may not seem that different, but they correspond to R2 values of 0.64 and 0.36, almost a factor of two difference in performance. Second, ρ does not care about the specific values predicted, only their relative order. As long as the best mutations tend to come out on top and the worst at the bottom, your model will get a high ρ score, even if the specific predictions are bad and the R2 is low. Some people may argue that for protein engineering or other applications of mutational effect prediction getting the relative ranking is good enough, and therefore using ρ is fine. But in my opinion, this is just an admission that the models aren’t very good yet, and that they tend to fail if we need more specific predictions than just relative ranks.
More from Genes, Minds, Machines
Limitations of protein language models applied to viral data

Current biological AI models don’t seem to work well for data from viral proteins. Specifically, I’m referring to protein language models applied to the problem of predicting effects of mutations. Protein language models are transformer-based AI models similar to ChatGPT but trained entirely on protein sequences. The most popular such models are
We still can’t predict much of anything in biology

AI has gotten amazingly good for programming. Claude Sonnet will zero- or one-shot small programming tasks without mistakes. And while I don’t think AI is ready to replace software engineers outright, or that vibe coding a fully featured app is a good idea, for simple tasks AI is outstanding. For example, I can perform basic data analysis, maybe visuali…
For simplicity, we refer to any measurable quantitative phenotype as “fitness.”
L. C. Vieira, S. Lin, C. O. Wilke (2026). Intrinsic dataset features drive mutational effect prediction by protein language models. bioRxiv. https://doi.org/10.64898/2026.03.08.710389
But note the performance of ESM C. It does surprisingly well on site-stratified data for cellular proteins, and extremely poorly on site-stratified data for viral proteins. ESM C is without doubt one of the best current pLMs, but only if you work with cellular proteins. For virus data, it is terrible.
For the full story, read the paper: https://doi.org/10.64898/2026.03.08.710389
We did the same for predicting antifungal resistance arising from mutations using ESM-2 embeddings and DMS data. When we split data by position performance dropped significantly as well but stayed above the random guess. But I didn't check the ratio of low- and high-variance sites in our data, it's a great idea, and I try to do it
Hm, wouldn’t splitting the training data by genes be even fairer? Train of 90% evaluate on the 10% held out? The promise of mutation prediction seems to be that we’d generalize the information captured in saturated mutagenesis models beyond the set of genes that have been studied? Would also be very interesting (but absolutely horrendously expensive, unless there are ESM snapshots partly through pretraining) to plot base training data volume vs R2, I think it might be related to “volume seen in base training”?