
We still can’t predict much of anything in biology
Biology is hard. Yes, even for AI.
AI has gotten amazingly good for programming. Claude Sonnet will zero- or one-shot small programming tasks without mistakes. And while I don’t think AI is ready to replace software engineers outright, or that vibe coding a fully featured app is a good idea, for simple tasks AI is outstanding. For example, I can perform basic data analysis, maybe visualize a dataset with a PCA or run a classifier, by sketching out what I want in a prompt and Claude will reliably write code that can do the task.1

It’s very tempting, in particular to the tech crowd, to look at this AI success in programming and extrapolate to other application areas. One popular area of extrapolation is biology. If we can teach an AI programming by feeding it millions of examples of code snippets, so the thinking goes, surely we can also teach it biology by feeding it millions of examples of biological data. And yes, to some extent this works. AlphaFold is pretty good.2 But, in practice, what happens more often than not, is not what I would call the AlphaFold experience.
This is my usual experience: I read about some new computational method that appears to work exceptionally well, I get excited because it’s exactly what I need for one of my projects, I try the method, and results are disappointing. Most of the time things don’t work, or at least not as well as expected. I have seen this play out so many times my default assumption is nothing is going to work. And anything that does actually work is a bit of a miracle. The only successful strategy is volume. If you’re trying sufficiently many things, some do in fact work, and those you can publish.
My latest example is with the software BindCraft, a tool built on top of AlphaFold to design peptide binders. Before I continue, let me emphasize that in my opinion BindCraft is really good. I have the utmost respect for its creators. It’s well documented, it’s easy to install, it does what it claims to do. Without doubt, it’s the best tool for designing peptide binders currently on the market. And yet, even though the authors write that they achieve “de novo protein binder design with experimental success rates of 10–100%,” in our own hands only maybe one out of about a hundred designs actually works. We can design binders with BindCraft, and we can design them more efficiently than using any other available method, but we still have to experimentally test hundreds of designs to get a handful of working inhibitors.3
This story is not unique to AI methods. I’ve been in computational biology for nearly three decades. Nothing has fundamentally changed during this entire time between how papers describe the success of new computational methods and how the methods actually perform in practice when you use them on your own system of interest. I remember, in the early 2000, David Baker was revolutionizing computational protein design with his Rosetta software suite, winning CASP competitions left and right, and writing papers that gave the impression computational protein design was solved. For example, computational design of novel folds was solved by 2003, protein docking was solved by 2003, enzyme design was solved by 2008, and atom-level co-folding of multiple peptide chains was solved by 2009. And yet, here we are, twenty years later, all of these topics are still active areas of research, and if you have any particular system of interest you may find that none of the available methods perform that well.
Now let me be absolutely clear: I’m not accusing anybody of faking data, doing sloppy science, or misrepresenting their results. All the papers I have cited here are examples of outstanding science practiced at the highest level. Instead, I think there are several things going on. First, biology is really difficult. Just because something works in one system doesn’t mean it’ll work in another. Second, there is publication bias. The examples that work get published, the ones that don’t work do not.4 Third, experienced PIs develop an instinctual understanding of which specific problems are amenable to their methodology. They may subconsciously or deliberately choose problems where the likelihood of success is high. I’m sure if you went to David Baker with a protein-design problem he could tell you right away whether you’re likely going to succeed or not and which software and parameter settings would maximize your chances. But, you’re not David Baker, you don’t have three decades of experience designing proteins, and so you’ll naively pick the wrong method or attempt a problem that is simply too hard. And the experience will be frustrating and it’ll feel like the available methods don’t perform anywhere near as well as the published papers suggest.
Going back to our experience with BindCraft, we’re starting to see patterns emerge of when it succeeds and when it fails. BindCraft appears to be much better at targeting certain binding pockets than others. You wouldn’t easily find this out from reading the paper, but generate a few thousand designs and you’re starting to get a feel for how the algorithm behaves. Similarly, there are non-obvious aspects to how you prepare your target structure that matter. In general, you have to make hundreds of seemingly inconsequential choices that can increase or decrease your chances of obtaining a successful binder design. Make all the right choices—and sprinkle in a bit of luck—and your design will work. Make a couple of wrong choices and it’ll fail.
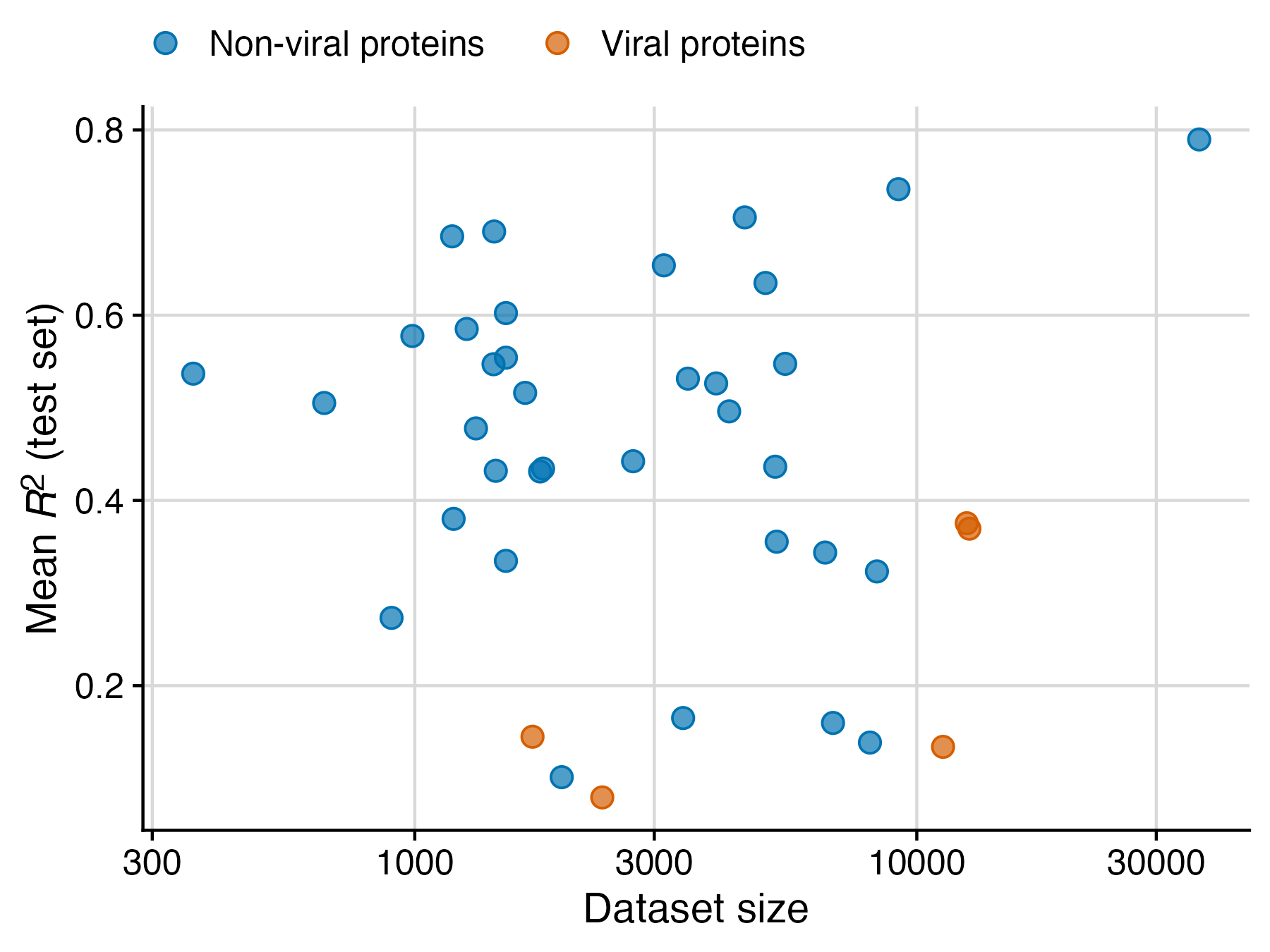
I want to emphasize that this post is not meant as a complaint about BindCraft. What I describe is a general phenomenon. For another example, look at my recent post about how protein language models fail on viral data.5 Specifically, look at the graph I included in that post. Even for the non-viral proteins, for at least half the datasets predictions mostly or entirely fail. An R2 below 0.5 is not a good prediction.
Why are predictions so poor? How can biology be so difficult? Biology is just physics and chemistry, and in principle we should be able to write down the equations of motion of any biological system and solve them numerically. In practice, however, any realistic biological system is way too large for this approach and we don’t have the required compute. It’s not feasible to rent an entire supercomputer for a year just to calculate the fitness effect of a single mutation. So we have to rely on shortcuts. The shortcuts that work are database lookups and interpolation. AlphaFold is a gigantic lookup and interpolation machine. It uses known structures in concert with covariation derived from multiple sequence alignments to link sequences to likely structures.6 Ask it to predict structures of novel folds or of sequences with no known homologs and its performance often craters. I’ve written previously about the limitations of AlphaFold. And a software such as BindCraft, built on AlphaFold, inherits all of AlphaFold’s limitations. To improve binder design, we first need to improve protein folding predictions.
And up to this point we’ve mostly talked about structure prediction. Move up one level to protein function and things get exponentially more difficult. Any given protein can have hundreds of different functions, when you consider all the different environments in which the protein can be expressed, different interaction partners it may come in contact with, and—for enzymes—different substrates it may act upon. If you wanted to construct a model that could reliably predict effects of mutations, you’d need it to achieve this task for every possible mutation in every single context in which the protein could occur. I hope you can see how big of a task this is, and, for AI approaches, how much data you’d need to collect to cover this enormous space evenly in your training set.
And once you’ve mastered protein function, you’re still only at the base of a gigantic mountain. First, there are molecules beyond proteins that also play important roles, such as RNA, DNA, lipids, glycans, various other small molecules, the list goes on and on. And second, you have to assemble individual molecules and their functions into pathways, and then into cells, and organ systems, and organisms, and from there on into populations and eventually ecosystems. You will encounter new challenges and complications at every new level of organization. You may hope that you can abstract away the lower levels as you move up in the hierarchy, but this will only get you so far. Individual genetic changes can have visible consequences at much higher levels of organization. As but one example, consider genetic variation linked to burrowing behavior in mice. In the end, all of biology is steered by what happens at the molecular level, and you can never quite get rid of the complexity and the unexpected effects or interactions.
I want to close on a more positive note. Even though computational predictions in biology are still fraught with failure, with every passing year science grinds forwards and we’re collectively accumulating knowledge. Our ability to manipulate biological systems is improving one step at a time. For example, we are much better today at designing peptide binders than we were twenty years ago. And at the same time, we still have many decades of work ahead of us. The world of biology is so large that complete mastery remains out of reach. Today we can design a car or an airplane entirely in silico, and when we build it it works on first try. Maybe, in a few decades, we’ll be able to do something comparable with a biological system.
Now I’d like to hear from you in the comments. Do you have similar experiences? Do your modeling or prediction efforts frequently fail? Or do you think I’m too negative and in your hands things generally work? In your mind, what’s the state of computational prediction in biology in 2025?
This post, or at least something like it, was requested by Eurydice.
In case you’re living under a rock and have no idea what I’m talking about, you can check out an example I have prepared here. In response to a simple prompt, Claude prepared a non-trivial piece of code that does exactly what I wanted it to do.
Just don’t go around saying AlphaFold has solved the protein-folding problem. It has not done this. There are many caveats and limitations. I have written about this previously.
We’re designing peptide binders as inhibitors of enzyme activity, so our actual problem is a bit more difficult than just binding, but that’s what real biology is like. You never actually want the exact same thing that was demonstrated in a paper. Your real-world use case is always a bit different, and typically more complicated.
There is little we can do about publication bias. Yes, negative results should be published, but you can’t publish every failed experiment. Most experiments fail for reasons that don’t warrant publication. Negative results are publishable when you are certain you have made a serious effort at producing the effect and it’s just not there. Negative results due to negligence, incompetence, or insufficient effort should not be published.
I know, I promised we’d write a paper on this. The paper is in the works. As always, things are more complicated than originally envisioned and take longer than expected.
And David Baker’s Rosetta software, which is not built on AI, is also fundamentally a lookup machine that stitches together motifs from known protein structures. All successful protein-folding methods rely on structure lookup and covariation analysis.
Great piece, Claus. Very much enjoying the rebirth of your blog.
"Biology is just physics and chemistry" -- you discuss the "levels of organization" later, but perhaps it's worth saying that "just" in the first quote is bearing more weight than is reasonable, and in the second quote, "levels" may have a more precise definition. There are layers of emergence between the physics/chemistry and the biology, that is, phenomena at some scale which are not reducible to features at smaller scales, even though the phenomena arise purely from smaller-scale features (consciousness from neurons, waterfalls from water molecules, murmurations from starlings, selective permeability from masses of lipids, etc.). As Phil Anderson pointed out, emergence acts as a kind of insulation between layers. So there is nothing to be gained by having a better theory of quantum mechanics when considering the behavior of starlings.
Lacking even any particularly useful theory of emergence, and confronted with the obvious wild variety of emergent phenomena, it seems likely that predictive theories of biology, which rests on a stack of emergence, are unknowably far off. Not that we won't have niche examples where we can do fairly well (cf. AlphaFold), but having an example in one niche helps us (where us includes AI) not at all with the next niche.
Interesting piece I certainly know what you mean and your point about Baker labs making it seemed like protein design was solved a decade ago was spot on i remember being confused about that and why we didn’t see more uses in real world.
I kind of have a totally opposite experience with neural network potentials though. These try to do exactly what you’re saying re simulating from the ground up. I routinely find them remarkably more capable than I would expect. For instance here I took one trained on crystal structures of small highly ordered periodic systems and used it to simulate the potassium ion channel and reproduced several known features + some new ones that are consistent with mutational studies. https://arxiv.org/abs/2411.18931 and here I simulate carbonic anhydrase where it also seemed to do a remarkably great job. ( https://arxiv.org/abs/2503.13789) Obviously still not perfect but the newer models are already a lot better than the one I used in those papers. I think their are at the right level of abstraction that the data is simple enough that it can be fully learnt but not so simple that it can be fitted l by hand.